 2024, Vol. 33
2024, Vol. 33
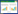




主动脉夹层(aortic dissection, AD)是心血管系统危急重症,Stanford A型者更为高危,可致心包填塞、心肌梗死等迅速导致死亡[1]。马凡综合征为因FBN1基因突变引起的少见的常染色体显性遗传病,可导致心血管系统等结缔组织脆弱性增加[2],由该病导致的主动脉夹层,病情进展更为迅速,救治难度更大。本文对2017年10月至2022年10月本院收治的7例马凡综合征导致Stanford A型主动脉夹层的患者的临床特点、治疗以及预后进行回顾性分析,总结临床经验,其中2例为首次报道的FBN1基因突变类型。
1 资料与方法 1.1 一般资料2017年10月至2022年10月就诊于温州医科大学附属东阳医院的全部7例马凡综合征致Stanford A型主动脉夹层的患者。男6例,女1例;年龄22~37岁,(30.4±5.3)岁,均因急性胸痛就诊,详细询问每位病人的家族史及遗传史,术前通过主动脉CTA、心动超声明确主动脉病变情况。除1例主动脉夹层远端终止于主动脉弓外,余6例均累及降主动脉。合并因主动脉夹层撕裂左、右冠状动脉开口导致心肌梗死1例,存在中度以上主动脉瓣关闭不全3例。1例既往因二尖瓣脱垂行二尖瓣机械瓣置换。
1.2 马凡综合征诊断方案根据2010年修订版Ghent方案进行马凡综合征诊断[3],主要指标有家族史、主动脉根部直径、晶状体脱位、FBN1基因突变、系统评分。系统评分共13项,包括:腕征和拇指征、胸廓畸形、足跟畸形、气胸史、腰骶部硬脊膜囊扩张、髋臼内陷、身体下部/上部或臂展/身高过大、脊柱侧凸或后凸、肘关节外展受限、面部特征、皮肤牵拉纹、严重近视、二尖瓣脱垂,总分超过7分即为系统评分阳性。
FBN1基因突变检测采用芯片捕获高通量测序,并Sanger法验证[4]。致病性判断根据修订版Ghent方案并结合文献报道等进行。
1.3 手术方法所有病例接受急诊手术,全身麻醉中度低温体外循环。先行右锁骨下动脉插管,再开胸,经右心房插入腔房管,阻断升主动脉后,切开升主动脉,探查主动脉根部病变情况,并经冠状动脉开口顺行灌注停跳液。右冠状动脉开口受累严重者则缝闭之,解剖出右冠状动脉近段,用大隐静脉行冠脉搭桥。先进行主动脉根部处理,本组患者皆存在严重主动脉根部病变且为青年患者,故采用带机械瓣的人工血管行Bentall术以置换主动脉根部。主动脉弓病变予孙氏手术[5],应用术中支架联合四分支人工血管于胸降主动脉内完成支架象鼻并置换主动脉弓,期间进行深低温停循环选择性脑灌注。主动脉重建完成后,开放升主动脉,心脏恢复搏动。吻合左锁骨下动脉、左颈总动脉、头臂干至四分支人工血管以重建之。主动脉根部外包自体主动脉壁并与右心房作一内引流。
2 结果 2.1 马凡综合征诊断情况7例患者根据2010年修订版Ghent方案都符合马凡综合征诊断。例1因合并严重脊柱侧突、胸廓畸形,且已植入二尖瓣机械瓣,未生育,父母健在。例2生育1子,8岁,存在漏斗胸等。例3生育1子1女,1子10岁,存在脊柱侧突等。例4母亲青年猝死,有一兄29岁,有气胸病史等。例5生育1女,13岁,存在二尖瓣脱垂等。例6母亲青年猝死,一位姨妈已患主动脉夹层。例7母亲青年猝死,母系亲属中2人已患主动脉夹层。故除例1外,其余6例判断存在家族史。所有患者都存在主动脉根部严重扩张,无合并眼晶状体脱位者,系统评分总分都为阳性,见表 1。
| 病例 | 性别 | 年龄(岁) | 家族史 | 主动脉根部直径(厘米) | 晶状体脱位 | 系统评分总分 |
| 1 | 女 | 31 | 无 | 5.8 | 无 | 13 |
| 2 | 男 | 32 | 有 | 6.7 | 无 | 7 |
| 3 | 男 | 35 | 有 | 5.1 | 无 | 8 |
| 4 | 男 | 25 | 有 | 6.2 | 无 | 12 |
| 5 | 男 | 37 | 有 | 5.6 | 无 | 10 |
| 6 | 男 | 22 | 有 | 6.1 | 无 | 10 |
| 7 | 男 | 31 | 有 | 6.0 | 无 | 7 |
例2、3、4、5进行了FBN1基因突变检测,都发现了致病突变,且例2之子、例3之子、例4之兄、例5之女都发现了与患者相同的致病突变,并有马凡综合征临床表现,例2、4为无义突变,见表 2、图 1。既往文献报道[6-7]明确为致病突变,故根据突变类型、家系验证、文献报道都确立了其致病性。
| 病例 | 核苷酸变化 | 氨基酸变化 | 突变类型 | 文献报道 | 家系验证 |
| 2 | c.1605dupA | p.Q536Tfs*8 | 框移突变 | 未报道 | 有 |
| 3 | c.4621C > T | p.Arg1541* | 无义突变 | 已报道 | 有 |
| 4 | c.200G > A | p.Cys67Tyr | 错义突变 | 未报道 | 有 |
| 5 | c.1090C > T | p.Arg364* | 无义突变 | 已报道 | 有 |

|
| A、B、C、D分别为例2、3、4、5 图 1 4例患者的FBN1基因测序峰图 |
|
|
例1因患者为二次开胸手术,既往因植入二尖瓣机械瓣而为华法林抗凝状态,合并严重脊柱侧突,且一般情况极差,仅行Bentall术,术后因呼吸衰竭而呼吸机脱机困难,后行气管切开,康复锻炼后顺利脱机、闭合气管切口。例2,开胸时死亡。例3、5、6行Bentall+孙氏手术。例4、7术中探查合并右冠状动脉开口撕脱,故行Bentall+孙氏手术+右冠状动脉旁路术。例4,首次手术后2个月因主动脉假性动脉瘤再次手术后死亡。例7,因术后出现严重低心排,后出现多脏器衰竭,于术后第3天死亡。即例1、3、5、6顺利度过围手术期并存活出院。
于2024年1月15日末次随访时,该4例仍存活,复查全主动脉CTA、心脏超声、心电图等,除例3存在主动脉根部轻度吻合口漏外,余3例康复情况良好,恢复日常生活。
3 讨论随着对其认识的深入和重视,医疗条件的改善,漏诊逐渐减少,主动脉夹层已经成为常见的心血管系统急重症。根据预后的巨大差异,Stanford分型简洁的根据是否累及升主动脉将主动脉夹层分为A、B型。其中A型者,因心包内的主动脉根部存在病变,而容易因急性心包积液出现心包填塞、冠状动脉口堵塞导致急性心肌梗死、主动脉瓣损害而引起急性关闭不全等原因而迅速导致病人死亡,故更应引起重视。
主动脉夹层常由未良好控制的高血压引起,由马凡综合征导致者,有其特殊性,既往报道不足。在一项纳入288例A型主动脉夹层的单中心回顾分析中,合并马凡综合征者为25例,即占8.68%[8],在本研究5年期间,本院共诊治A型主动脉夹层82例,即马凡综合征病例占8.54%,与前报道占比类似。故需加强对于马凡综合征的认识,以减少误诊、漏诊。
马凡综合征患者发生主动脉夹层时较一般人群年轻,本组发生主动脉夹层时平均年龄仅30岁,与一般人群发生主动脉夹层年轻约10~20岁[9]。
马凡综合征患者发生主动脉夹层常不合并高血压,其病因为为马凡综合征致病的FBN1基因突变,导致全身结缔组织的原纤维蛋白1异常,引起包括心血管系统等脆弱性增加所致。
马凡综合征患者发生主动脉夹层通常在其逐渐进展的主动脉窦部扩张的基础上发生,其主动脉窦显著扩大,而升主动脉直径可基本正常[10],在阅读CT图像时应注意此特点,且因主动脉窦扩张严重,其主动脉根部病变常非常严重,容易出现冠状动脉开口撕脱、主动脉瓣畸形关闭不全等严重心脏并发症而导致患者迅速死亡。
另需重视马凡综合征的其他特点[11]:在病史询问上,要注意家族史,应详细询问所有血亲的疾病和存活情况,尤其要重视猝死病史;既往史重点抓住二尖瓣病变、气胸史等;脊柱畸形、胸廓畸形等骨骼病变、皮肤牵拉纹等体征可非常显著,注意识别;腰骶部硬脊膜囊扩张、髋臼内陷可在CT上有特征性影像;眼晶状体脱位虽为重要的特征性病变,但本组病例为主动脉夹层就诊,无发生者,但仍需重视该特征。通过提高马凡综合征的识别率,进而减少主动脉夹层等严重病情的漏诊。
本组7例中,有4例因患方有进一步明确诊断、明确家系中的其他成员的FBN1基因型、避免遗传至下一代等意愿,故予行基因诊断。其中例3、例5为无义突变,导致该FBN1基因处密码子变异为终止密码子,引起所编码的原纤维蛋白1被截短,致严重功能障碍导致发病,这与既往报道相符[12]。例2为框移突变,例4为错义突变,为2种既往未见文献报道的FBN1致病突变,丰富了对于马凡综合征和FBN1基因的认识。
Stanford A型主动脉夹层因病情凶险,一经诊断即应开胸手术抢救。本组马凡综合征导致的Stanford A型主动脉夹层患者病情严重,例2因存在左右冠状动脉开口严重撕脱而致大面积心肌梗死,开胸时即死亡,例4与例7也存在右冠状动脉开口受累,虽度过手术,但仍分别因主动脉假性动脉瘤和低心排而死亡。余4例手术治疗效果良好,但因马凡综合征者主动脉再次出现病变可能性相对大,目前仍在继续随访中。
利益冲突 所有作者声明无利益冲突
| [1] | 中国医师协会心血管外科分会大血管外科专业委员会. 主动脉夹层诊断与治疗规范中国专家共识[J]. 中华胸心血管外科杂志, 2017, 33(11): 641-654. DOI:10.3760/cma.j.issn.1001-4497.2017.11.001 |
| [2] | 谢恩泽华, 公兵, 赵锐, 等. 马方综合征发病机制的研究进展[J]. 中华心血管病杂志, 2019, 47(8): 664-668. DOI:10.3760/cma.j.issn.0253-3758.2019.08.015 |
| [3] | Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome[J]. J Med Genet, 2010, 47(7): 476-485. DOI:10.1136/jmg.2009.072785 |
| [4] | 杨明烽, 楼杨勇, 吕小辉, 等. 马方综合征临床表现及FBN1基因检测5例分析[J]. 浙江医学, 2018, 40(15): 1736-1738. DOI:10.12056/j.issn.1006-2785.2018.40.15.2017-2142 |
| [5] | 中国医药教育协会心脏外科分会及中国医药生物技术协会心血管外科技术与工程分会主动脉术式专家共识编写组. 主动脉术式中国专家共识: 孙氏手术[J]. 中华胸心血管外科杂志, 2021, 37(5): 270-273. DOI:10.3760/cma.j.cn112434-20210223-00068 |
| [6] | Baetens M, van Laer L, De Leeneer K, et al. Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes[J]. Hum Mutat, 2011, 32(9): 1053-1062. DOI:10.1002/humu.21525 |
| [7] | Tjeldhorn L, Rand-Hendriksen S, Gervin K, et al. Rapid and efficient FBN1 mutation detection using automated sample preparation and direct sequencing as the primary strategy[J]. Genet Test, 2006, 10(4): 258-264. DOI:10.1089/gte.2006.258-264 |
| [8] | 肖子亚, 姚晨玲, 顾国嵘, 等. 580例主动脉夹层患者临床特征及预后分析[J]. 中华急诊医学杂志, 2016, 25(5): 644-649. DOI:10.3760/cma.j.issn.1671-0282.2016.05.020 |
| [9] | Milleron O, Arnoult F, Delorme G, et al. Pathogenic FBN1 genetic variation and aortic dissection in patients with Marfan syndrome[J]. J Am Coll Cardiol, 2020, 75(8): 843-853. DOI:10.1016/j.jacc.2019.12.043 |
| [10] | Chung JC. Pathology and pathophysiology of the aortic root[J]. Ann Cardiothorac Surg, 202, 12(3): 159-167. DOI:10.21037/acs-2023-avs1-17 |
| [11] | Chiu HH. An update of medical care in Marfan syndrome[J]. Tzu Chi Med J, 2021, 34(1): 44-48. DOI:10.4103/tcmj.tcmj_95_20 |
| [12] | Baudhuin LM, Kotzer KE, Lagerstedt SA. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events[J]. Genet Med, 2015, 17(3): 177-187. DOI:10.1038/gim.2014.91 |